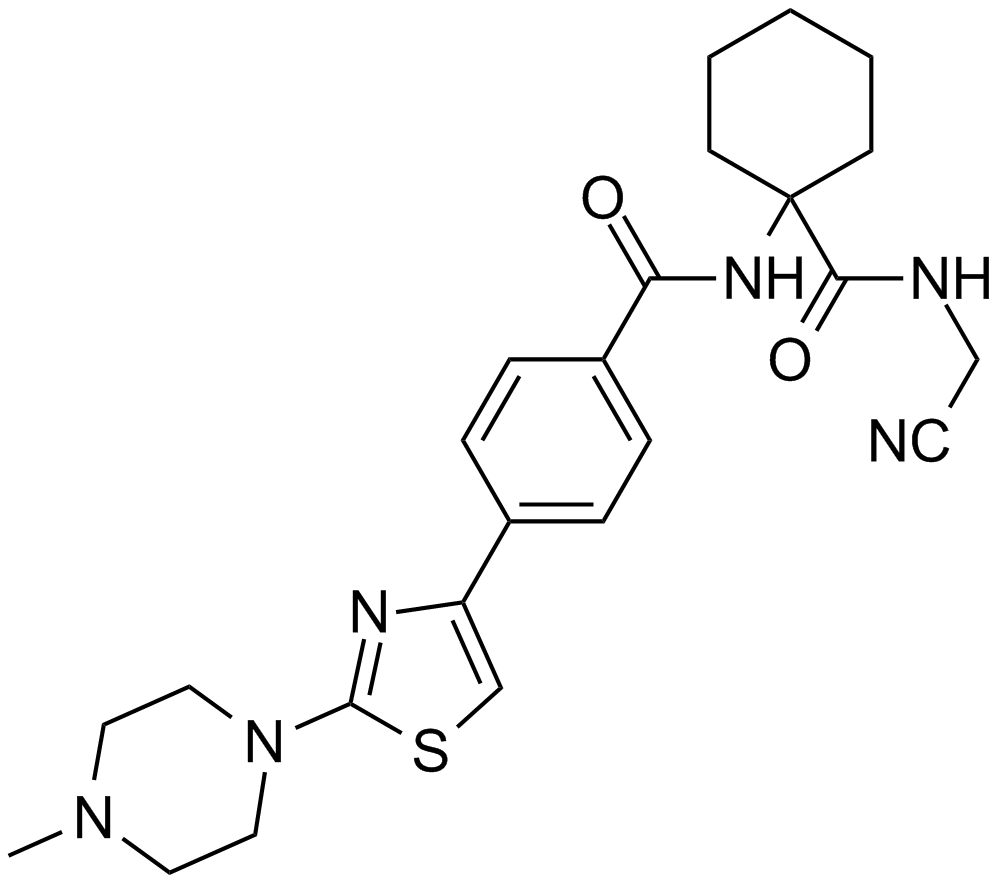
L 006235

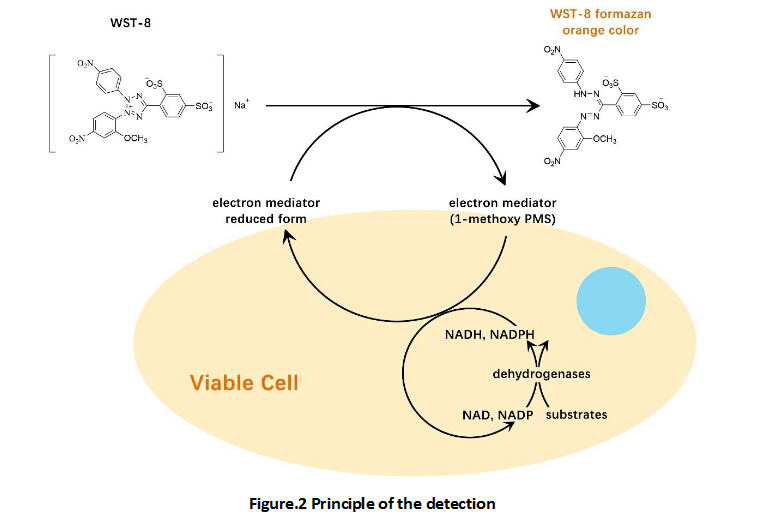
IC50: 0.25 nM
L-006235 is a potent and selective inhibitor of Cathepsin K.
In vitro: After dilution of L-006235 to 0.05 nM, the cathepsin K enzyme activity was initially inhibited, but slowly recovered with a first-order rate constant of 0.023 s-1. The final steady-state enzyme activity was 80-90% that of control, suggesting the complete reversibility of the L-006235-cathepsin K complex. L-006235 was found to be not a substrate for the nitrilase activity of Cat K [1].
In vivo: L-006235 was orally bioavailable in rats, with a terminal half-life of over 3 h. L-006235 was orally dosed in ovariectomized rhesus monkeys once per day for 7 days. Results showed that collagen breakdown products were dose-dependently reduced by up to 76%. Plasma concentrations of L-006235 above the bone resorption IC50 after 24 h indicated a correlation between functional cellular and in vivo assays. These findings suggested that the inhibition of collagen breakdown by cathepsin K inhibitors, such as L-006235, might be useful in osteoporosis and other indications involving bone resorption [1].
Clinical trial: N/A
Reference:
[1] Palmer JT,Bryant C,Wang DX et al. Design and synthesis of tri-ring P3 benzamide-containing aminonitriles as potent, selective, orally effective inhibitors of cathepsin K. J Med Chem.2005 Dec 1;48(24):7520-34.
| Physical Appearance | White solid |
| Storage | Store at 4°C |
| M.Wt | 466.6 |
| Cas No. | 294623-49-7 |
| Formula | C24H30N6O2S |
| Solubility | <46.66mg/ml in DMSO |
| Chemical Name | N-[1-(cyanomethylcarbamoyl)cyclohexyl]-4-[2-(4-methylpiperazin-1-yl)-1,3-thiazol-4-yl]benzamide |
| SDF | Download SDF |
| Canonical SMILES | CN(CC1)CCN1c1nc(-c(cc2)ccc2C(NC2(CCCCC2)C(NCC#N)=O)=O)c[s]1 |
| Shipping Condition | Small Molecules with Blue Ice, Modified Nucleotides with Dry Ice. |
| General tips | We do not recommend long-term storage for the solution, please use it up soon. |
| Potent, reversible cathepsin K inhibitor (IC50 = 0.25 nM) that displays > 4000-fold selectivity over cathepsins B, L and S. | ||||||
| Targets | cathepsin K | |||||
| IC50 | 0.25nM | |||||
Quality Control & MSDS
- View current batch:
-
Purity = 98.00%
- COA (Certificate Of Analysis)
- MSDS (Material Safety Data Sheet)
- Datasheet
Chemical structure