

How to Request a Quote
Form
Please download and fill in the

Email
And then send the form to [email protected]
Feedback
We will contact you by email as soon as possible, please make sure your Email address is correct and check your email in time.
1. Overview
Microorganisms are the most widespread organisms on the earth. Over 99% of natural microorganisms cannot be isolated and cultured clonally, while traditional isolation and culture-dependent methods challenge the study of microorganisms in their natural environment. Based on microbial characteristics,researchers developed several culture-free ways to explore the diversity structure and functional genome of microorganisms. Metagenomics is one of them.
Unlike 16S/18S/ITS Sequencing, which only targets specific microorganisms, metagenomic research takes all microorganism genomes in the environment as the research objects. The metagenomic service provided by APExBIO uses high-throughput sequencing of whole-genome DNA in environmental samples to obtain the data of a single sample. Then, based on de novo assembly, analyze the diversity of the microbial community structure, the genetic composition of the microbial population, and the metabolic pathways related to the specific environment, to further explore valuable genes and interrelationships within the microbial community.2. Workflow
2.1 Library preparation & Sequencing
When we received your sample(s), the sample quality test will be conducted first. Then we use qualified DNA sample(s) to construct library(s). After that, we will conduct library quality test. And finally, the qualified library(s) will be used for sequencing (Illumina PE150). The bioinformatics analysis will be carried on with sequencing data (Raw data). In order to provide high-quality data for you, each step of sample testing, library construction and sequencing is strictly controlled by APExBIO.
Our experimental procedures are shown in Figure2.1.
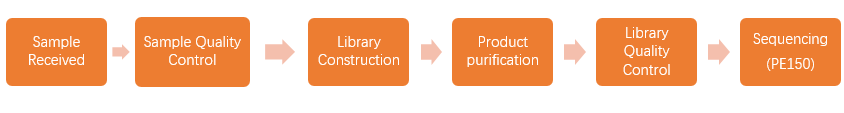
Figure 2.1 Experimental Procedures
2.2 Bioinformatics Analysis
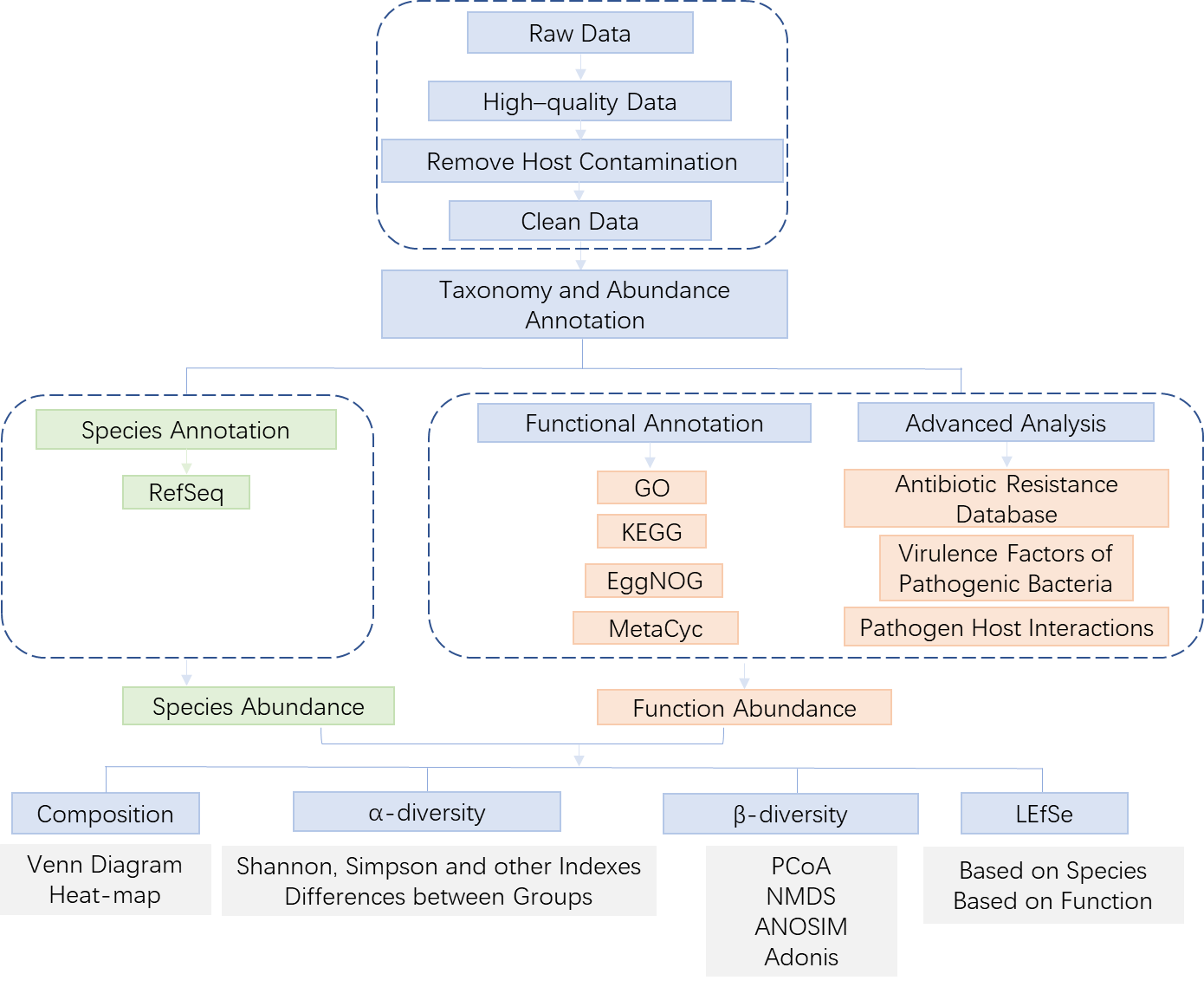
Figure 2.2.0 Bioinformatics Analysis Workflow
3. Applications
3.1 The Alterations of Vaginal Microbiome in HPV16 Infection as Identified by Shotgun Metagenomic Sequencing
3.1.1 Abstract
The association of microbiome imbalance with cancer development is being one of the research hotspots. Persistent HPV infection is a causal event in cervical cancer initiation, but, little is known about the microbiome composition and function in HPV infection. Here we identified the compositional and functional alterations on vaginal samples from 27 HPV16 positive women and 25 age-matched HPV negative controls using shotgun metagenomic sequencing, to provide a comprehensive investigation describing the microbial abundances and enriched metabolic functions in cervicovaginal metagenomes. We further employed qPCR assays to evaluate two selected gene markers of HPV16 infection in an independent validation cohort consisting of 88 HPV16 positive women and 81 controls, and six selected species markers in a subset of validation cohort of 45 HPV16 positive women and 53 controls. We found that the relative abundance of dominant Firmicutes was lower, Actinobacteria, Fusobacteria and viruses phyla were significantly higher in the HPV16-positive group; 77 genera including Gardnerella, Peptostreptococcus, and Prevotella were higher, and 20 genera including Lactobacillus and Aerococcus were lower in the HPV16-positive women. Abundance of 12 genes, 17 genera, and 7 species biomarkers showed an excellent predictive power for the HPV16-positive individuals, with 0.861, 0.819, and 0.918, respectively, of the area under the receiver-operating characteristic curve (AUC). We further characterized the microbial function, and revealed that HPV16-positive women were enriched in metabolism and membrane transport, and depleted by glycan biosynthesis and metabolism, and replication and repair. Quantitative PCR measurements validated that one gene marker and three species were significantly enriched in HPV16-positive women. These results highlight a fundamental fact that there are altered composition and function of the vaginal microbiome in HPV16-positive women, suggesting that vaginal dysbiosis may be associated with HPV infection in the female genital tract.
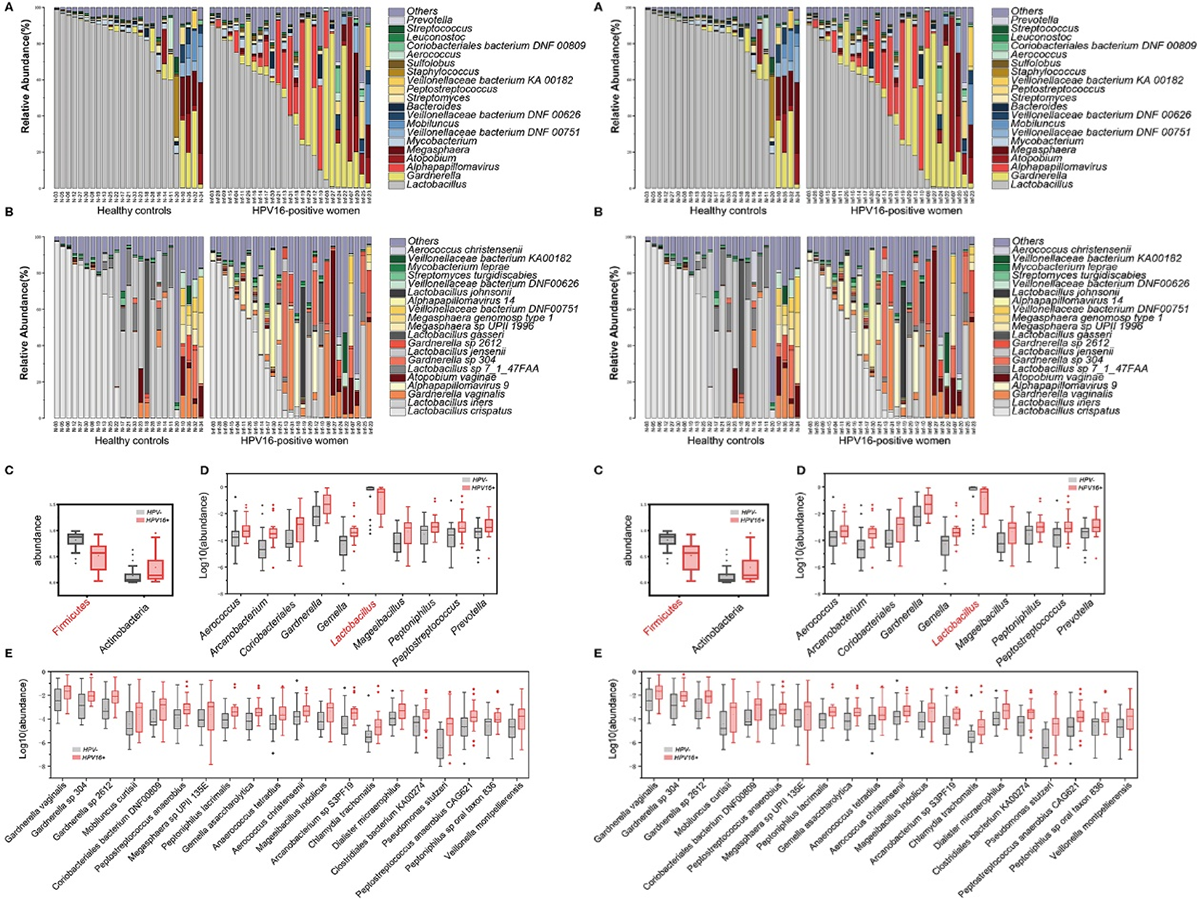
Figure 3.2.1 Phylogenetic profiles in vaginal microbes between HPV16-positive women and controls. Composition of vaginal microbiota in two groups at the genus level (A) and species level (B). Comparison of differentially abundant phylotypes identified by the Wilcoxon rank-sum test, at phyla (C), genera (D), and species (E) level, respectively. Only the top 2 phyla, top 10 genera and top 20 species are shown. The phylotypes enriched in the control group are colored with red. The relative abundances are shown by boxplot. Boxes represent the interquartile ranges, lines inside the boxes denote medians, and “+” denotes means.
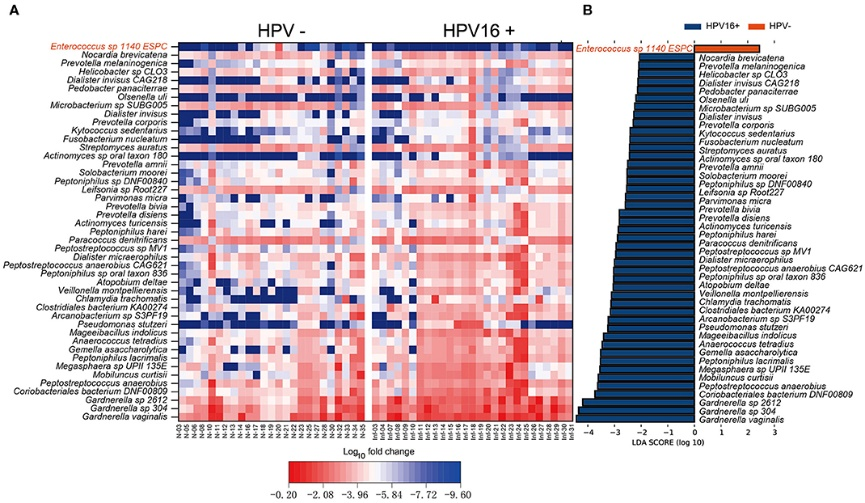
Figure 3.2.2 Vaginal microbiome as HPV16-infection markers. (A) Heatmap showing the relative fold change of bacterial species in HPV16 infection. The species enriched in controls are colored with red. (B) Histogram of the LDA scores computed for species differentially abundant between HPV16-positive women and controls. The LDA scores (log10) > 2 are listed.
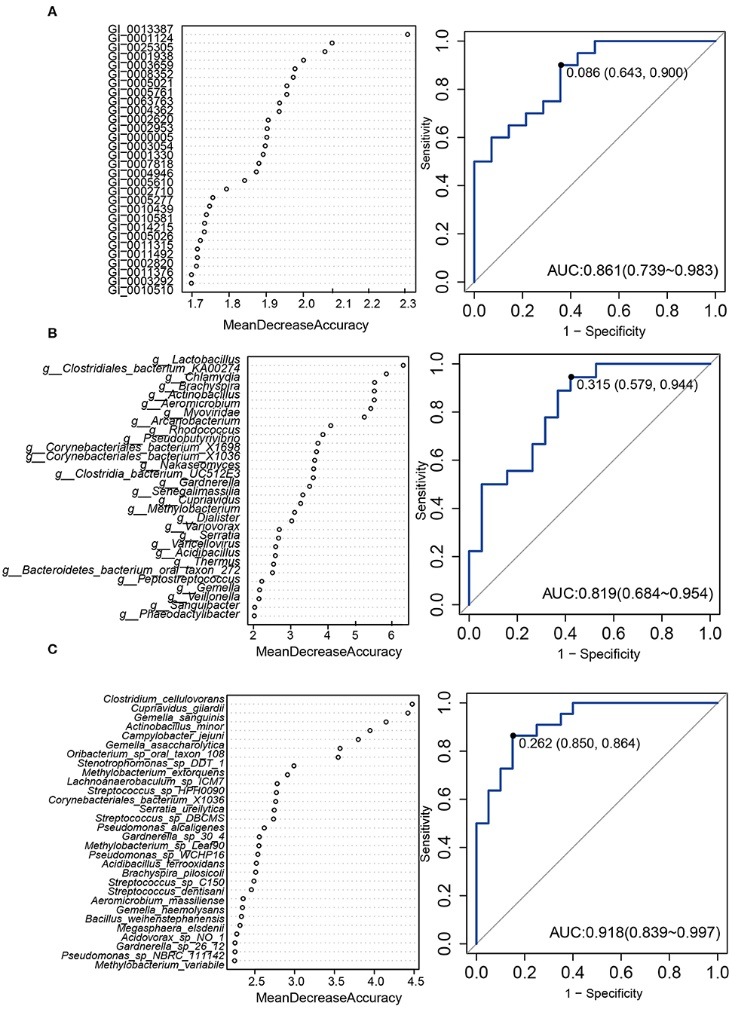
Figure 3.2.3 A predictive model of importance based on the gene/genus/species-level abundance profile using random forests (RF). The relative importance of each gene (A)/genus (B)/species (C) in the predictive random forest model using the mean decreasing accuracy. ROC curve generated by the RF using 12 genes (A)/17 genus (B)/7 species (C) in the vaginal microbiota. The plots shown in the ROC represent the corresponding optimal threshold.
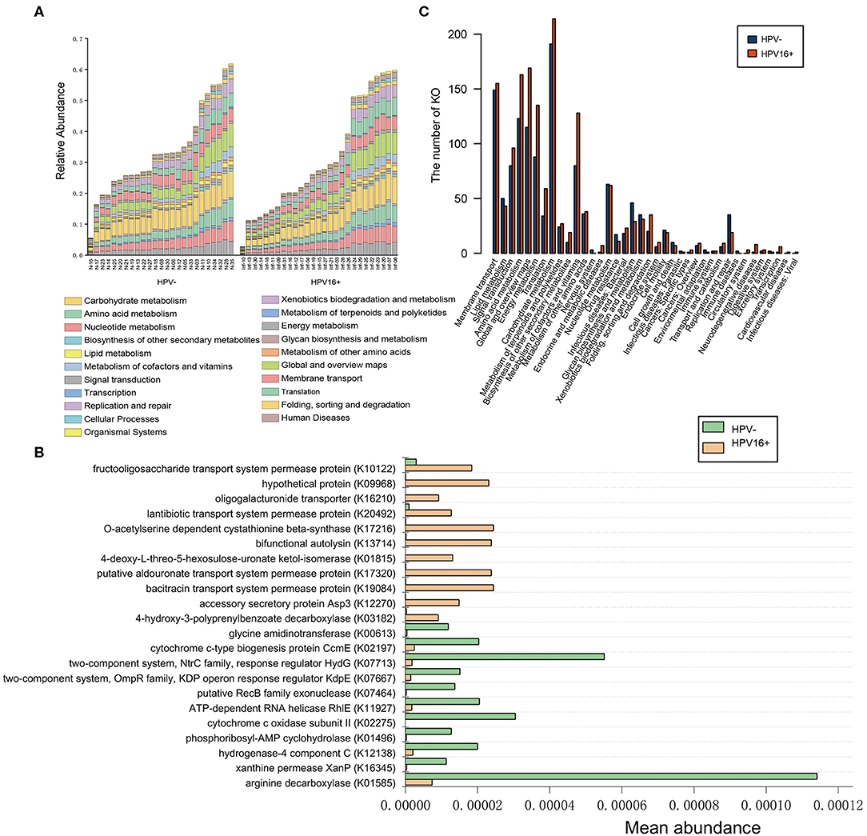
Figure 3.2.4 Functional predictions for the vaginal microbiome of the HPV16-positive and control groups. (A) The abundance of each sample at level 2 metabolic pathway. (B) The KOs with significantly different abundances in the vaginal microbiome identified by Metastats analysis (P, FDR < 0.05). (C) Comparison between the HPV16-positive group-enriched and the control-enriched KO markers on level 2 of the KEGG functional category.
4. References
[1] Yang Q, Wang Y, Wei X, Zhu J, Wang X, Xie X and Lu W (2020) The Alterations of Vaginal Microbiome in HPV16 Infection as Identified by Shotgun Metagenomic Sequencing. Front. Cell. Infect. Microbiol. 10:286. doi: 10.3389/fcimb.2020.00286