
Search results for: 'alzheimer'
-
 A1038 Amyloid β-Peptide (10-20) (human)Summary: Initiates neurodegeneration in Alzheimer disease
A1038 Amyloid β-Peptide (10-20) (human)Summary: Initiates neurodegeneration in Alzheimer disease -
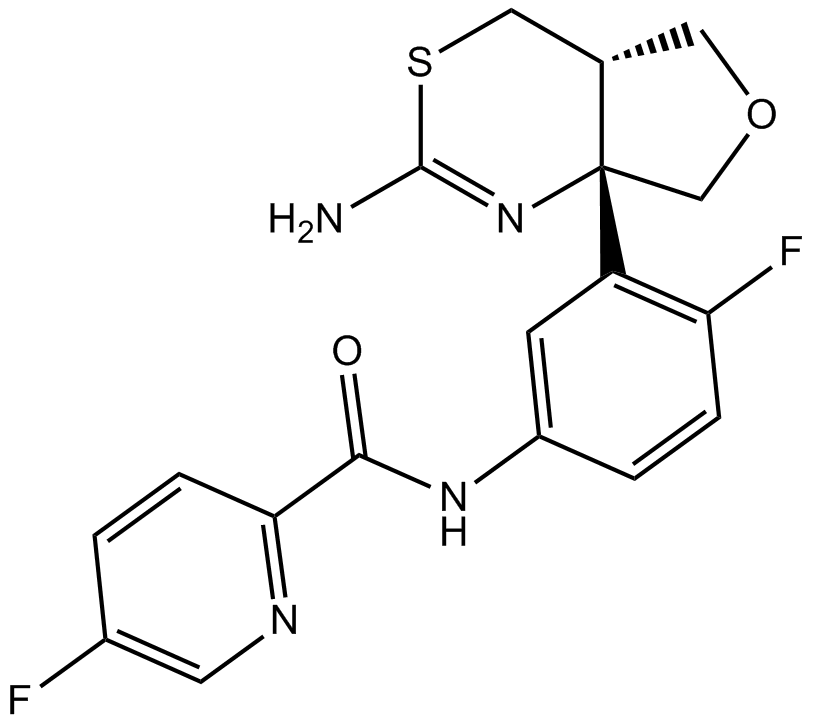 A8465 LY28867211 CitationTarget: BACESummary: BACE inhibitor
A8465 LY28867211 CitationTarget: BACESummary: BACE inhibitor -
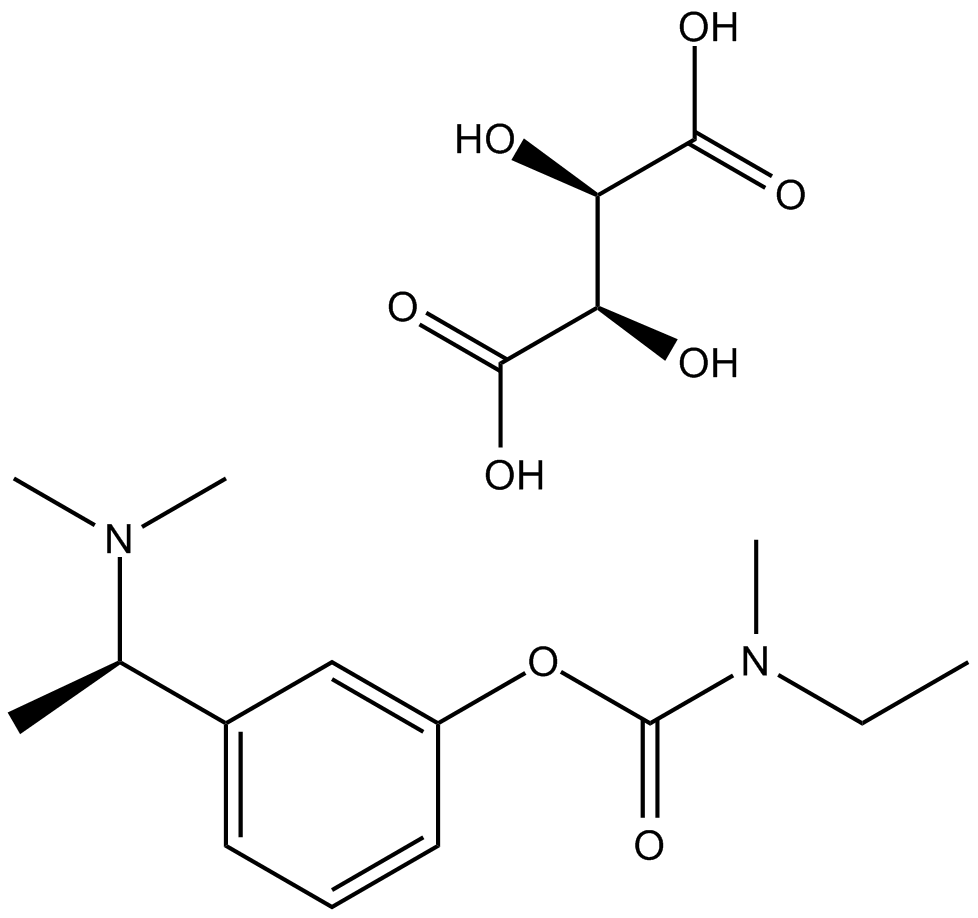 A8515 Rivastigmine TartrateSummary: AChR inhibitor
A8515 Rivastigmine TartrateSummary: AChR inhibitor -
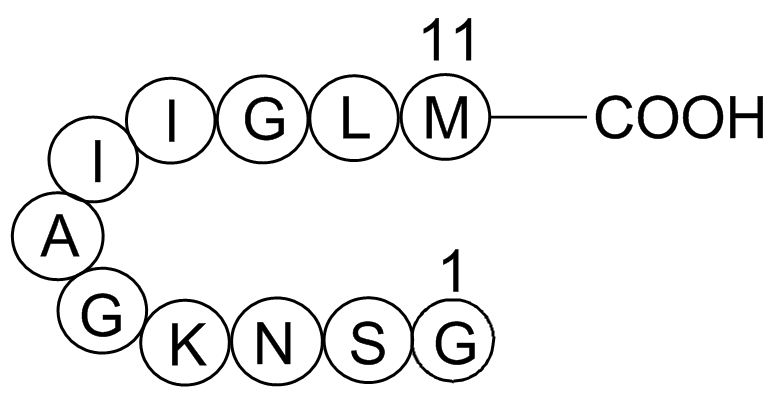 A1039 Amyloid Beta-peptide (25-35) (human)1 CitationSummary: β-amyloid peptide fragment; for Alzheimer’s disease-related neurotoxicity models
A1039 Amyloid Beta-peptide (25-35) (human)1 CitationSummary: β-amyloid peptide fragment; for Alzheimer’s disease-related neurotoxicity models -
 BA8516 CAD031Summary: CAD031 is a derivative of the Alzheimer's disease (AD)-targeting agent J147 with neuroprotective and memory-enhancing properties.
BA8516 CAD031Summary: CAD031 is a derivative of the Alzheimer's disease (AD)-targeting agent J147 with neuroprotective and memory-enhancing properties. -
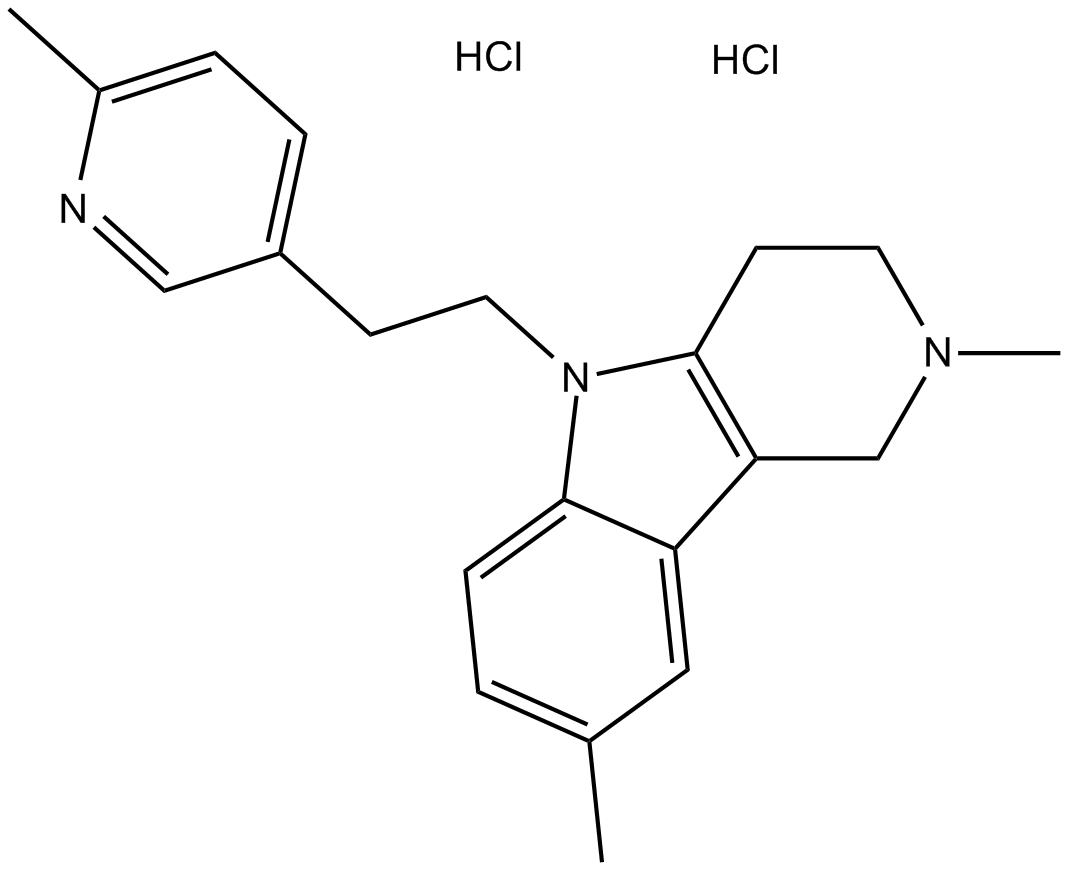 B1572 LatrepirdineTarget: Voltage-gated Calcium Channels (CaV)|NMDA receptor|adrenergic receptor|Histamine receptors|GluR|5-HT receptor|dopamine receptors|AMPA receptorSummary: Brain cell death inhibitor
B1572 LatrepirdineTarget: Voltage-gated Calcium Channels (CaV)|NMDA receptor|adrenergic receptor|Histamine receptors|GluR|5-HT receptor|dopamine receptors|AMPA receptorSummary: Brain cell death inhibitor -
 C8646 β-Amyloid (1-16)Summary: β-Amyloid (1-16) is a metal-binding peptide fragment derived from β-amyloid, associated with Alzheimer's disease pathology.
C8646 β-Amyloid (1-16)Summary: β-Amyloid (1-16) is a metal-binding peptide fragment derived from β-amyloid, associated with Alzheimer's disease pathology. - BA8408 AzeliragonSummary: Azeliragon (TTP488) is a bioavailable late-stage glycosylation end product receptor inhibitor that mitigates the progression of mild Alzheimer's disease (AD).
- BA8426 OAB-14Summary: OAB-14, a Bexarotene derivative, ameliorates Alzheimer's disease-associated pathology and cognitive deficits by increasing beta-amyloid clearance in APP/PS1 mice.
-
 K2242 Fluorometric Proteasome 20S Activity Assay KitSummary: A kit for the detection of proteasome 20S activity by the fluorescent substrate LLVY-R110
K2242 Fluorometric Proteasome 20S Activity Assay KitSummary: A kit for the detection of proteasome 20S activity by the fluorescent substrate LLVY-R110