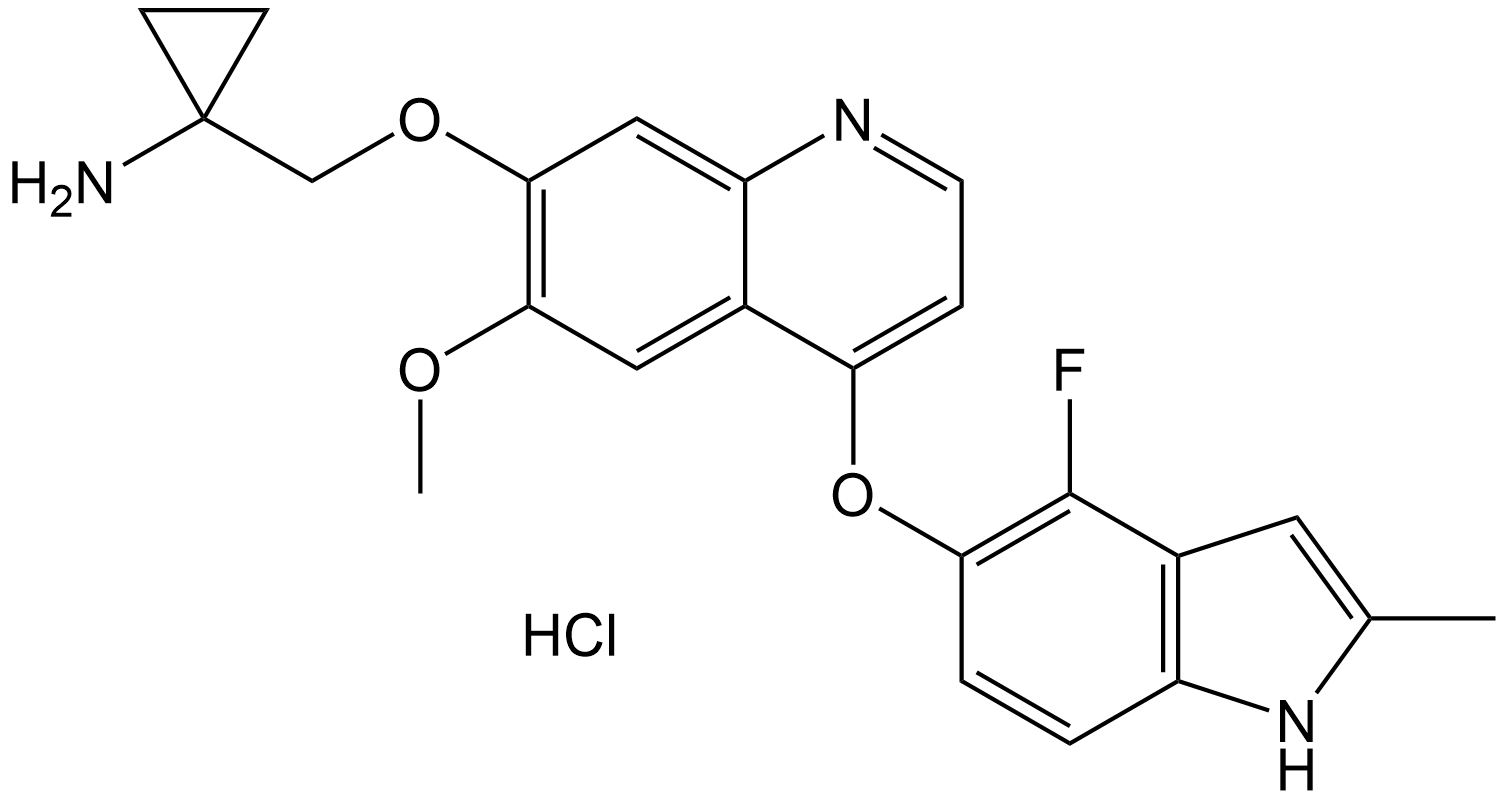
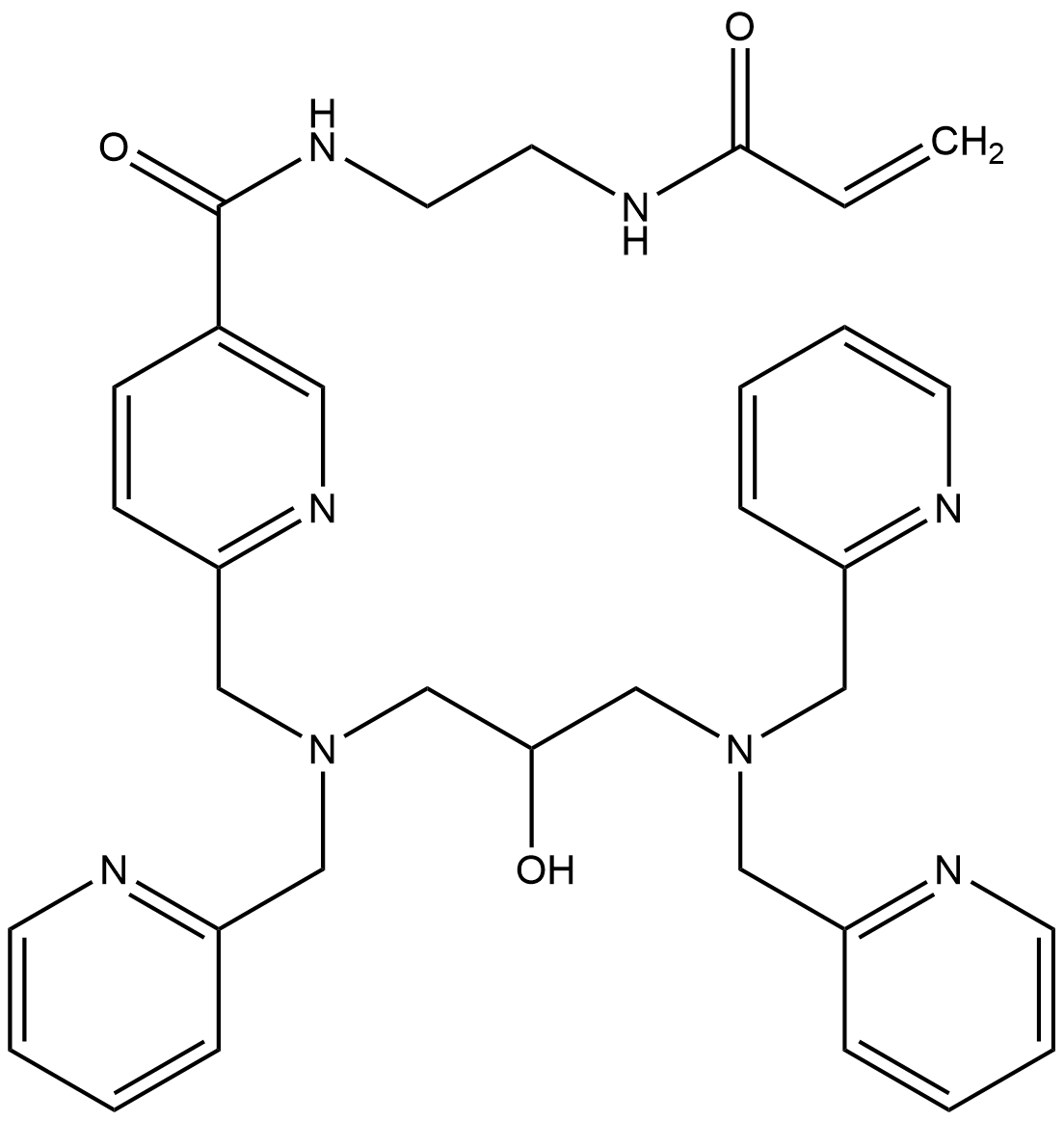
Anlotinib (hydrochloride)
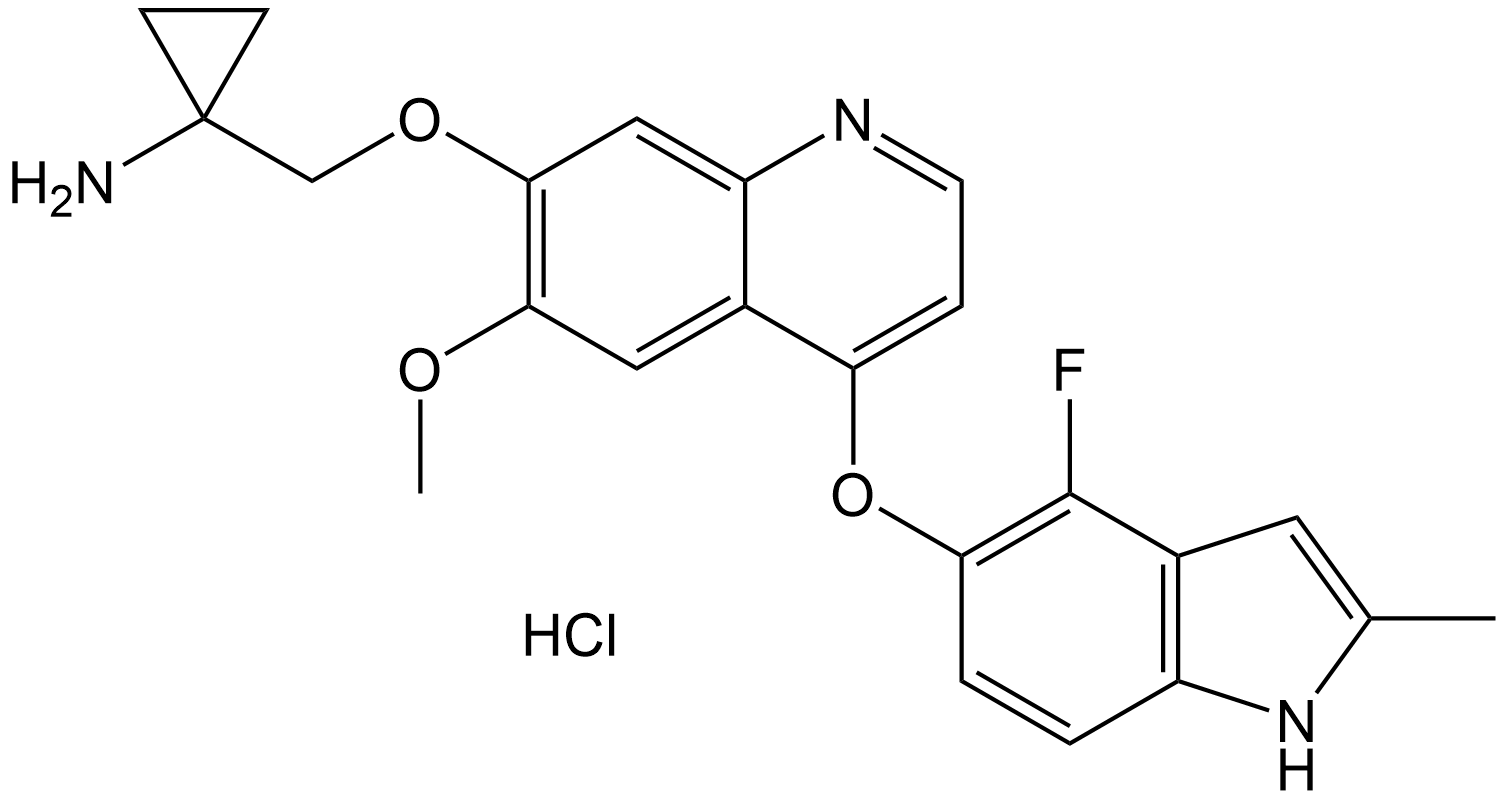
Anlotinib hydrochloride (CAS 1058157-76-8) is a novel small-molecule multi-target tyrosine kinase inhibitor (TKI).
In the cellular-stage studies using human vascular endothelial cells (EA.hy 926), the core focus was on the anti-angiogenic activity and mechanism of action of anlotinib. Its key targets are VEGFR2, PDGFRβ, FGFR1, and the downstream ERK signaling pathway. The experiments used doses of 0.01 μM, 0.1 μM, and 1 μM (a concentration range with no obvious effect on cell viability; the half-maximal inhibitory concentration IC₅₀ for cell viability was 30.26 μM). The results showed that anlotinib displayed significantly stronger inhibitory activity against PDGFRβ (IC₅₀ = 8.7 ± 3.4 nM) and FGFR1 (IC₅₀ = 11.7 ± 4.1 nM) than sunitinib (Cat. No.: B1045) and sorafenib (Cat. No.: A3009). Its inhibitory activity against VEGFR2 (IC₅₀ = 5.6 ± 1.2 nM) was second only to sunitinib and far superior to sorafenib. Anlotinib could inhibit VEGF/PDGF-BB/FGF‑2–induced cell migration (with a maximum inhibition rate of 75.32% at 1 μM) and capillary-like tube formation in a concentration-dependent manner. It also significantly inhibited the phosphorylation of the three core targets (with maximum inhibition rates of 56.37%, 41.1%, and 45.0% at 1 μM, respectively) and blocked ERK signal transduction. Overall, its effect was superior to that of three clinically used agents: sunitinib, sorafenib, and nintedanib (Cat. No.: A8252).
In animal-stage studies, anlotinib hydrochloride was investigated mainly in rats, tumor-bearing mice, and dogs. In addition, rat aortic ring and chick embryo chorioallantoic membrane (CAM) models were used for validation to comprehensively explore its pharmacokinetic and disposition characteristics, in vivo anti-angiogenic effects, and safety, supplemented by in vitro assessments to clarify its potential for drug–drug interactions. The compound shows good membrane permeability and is rapidly absorbed after oral administration. The oral bioavailability is 28%–58% in rats and 41%–77% in dogs, and the terminal half-life in dogs (22.8 ± 11.0 h) is significantly longer than in rats (5.1 ± 1.6 h), mainly due to the difference in total plasma clearance between the two species (5.35 ± 1.31 L·h⁻¹·kg⁻¹ in rats vs 0.40 ± 0.06 L·h⁻¹·kg⁻¹ in dogs). In terms of distribution, the apparent volume of distribution is relatively large in both rats (27.6 ± 3.1 L/kg) and dogs (6.6 ± 2.5 L/kg), and plasma protein binding is high (97% in rats, 96% in dogs, 93% in humans). In humans, anlotinib mainly binds to albumin and lipoproteins. In rats and tumor-bearing mice, the concentrations in tissue homogenates are markedly higher than the corresponding plasma concentrations; in rats, exposure is relatively high in the lung, liver, and kidney, and in tumor-bearing mice, the tumor tissue concentration can reach up to 13 times that of plasma.
Regarding metabolism, cytochrome P450–mediated oxidation is the main elimination pathway, with human CYP3A exhibiting the strongest metabolic capacity. Multiple metabolites, including hydroxylated and dealkylated products, can be detected in rats and dogs, while the proportion of unchanged parent drug excreted in urine, feces, and bile is low. For anti-angiogenic effects in vivo, the targets are consistent with those in the cellular studies: VEGFR2, PDGFRβ, FGFR1, and the downstream ERK pathway. In the rat aortic ring assay, doses of 0.01 μM, 0.1 μM, and 1 μM were used; in the CAM model, doses of 0.3 nmol/CAM, 1 nmol/CAM, and 1.5 nmol/CAM were applied. The results showed that anlotinib inhibited microvessel sprouting in rat aortic rings and neovascular density in the CAM model in a concentration-dependent manner, with the inhibition rate at 1 μM being 15%–30% higher than that of the control drug in the aortic ring assay, and the inhibition rate at 1.5 nmol/CAM being significantly higher than that of sunitinib and other comparators.
In terms of safety, the 14‑day oral median lethal dose (LD₅₀) reached 1735.9 mg, far higher than the clinical therapeutic dose, and systemic toxicity was mild, with no obvious liver, kidney, or bone marrow damage and no reproductive or genetic toxicity. Regarding drug–drug interactions, anlotinib shows some in vitro inhibitory effects on CYP3A4 and CYP2C9 (IC₅₀ < 1 μmol/L), but based on early human pharmacokinetic data, the associated risk is considered low, and the overall safety profile is favorable.
References:
[1] Lin B, Song X, Yang D, Bai D, Yao Y, Lu N. Anlotinib inhibits angiogenesis via suppressing the activation of VEGFR2, PDGFRβ and FGFR1. Gene. 2018 May 15;654:77-86. doi: 10.1016/j.gene.2018.02.026. Epub 2018 Feb 14. Erratum in: Gene. 2020 Jan 10;723:144119. doi: 10.1016/j.gene.2019.144119. PMID: 29454091.
[2] He C, Wu T, Hao Y. Anlotinib induces hepatocellular carcinoma apoptosis and inhibits proliferation via Erk and Akt pathway. Biochem Biophys Res Commun. 2018 Sep 18;503(4):3093-3099. doi: 10.1016/j.bbrc.2018.08.098. Epub 2018 Aug 23. PMID: 30146257.
| Storage | Store at -20°C |
| M.Wt | 443.9 |
| Cas No. | 1058157-76-8 |
| Formula | C23H23ClFN3O3 |
| Chemical Name | 1-(((4-((4-fluoro-2-methyl-1H-indol-5-yl)oxy)-6-methoxyquinolin-7-yl)oxy)methyl)cyclopropan-1-amine hydrochloride |
| Canonical SMILES | COC1=C(OCC2(CC2)N)C=C3C(C(OC(C=CC4=C5C=C(C)N4)=C5F)=CC=N3)=C1.Cl |
| Shipping Condition | Small Molecules with Blue Ice, Modified Nucleotides with Dry Ice. |
| General tips | We do not recommend long-term storage for the solution, please use it up soon. |
|
Cell experiments: |
|
|
Cell lines |
EA.hy 926 |
|
Cell culture |
The cells were routinely cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 U/mL streptomycin, and subcultured in a humidified incubator at 37°C with 5% CO₂. |
|
Reaction condition |
1. Cell Viability Assay: The cells were seeded at a density of |
|
Results |
The half-maximal inhibitory concentration (IC₅₀) of anlotinib against EA.hy 926 cells at 24 h was 30.26 μM. Anlotinib at concentrations of 0.01 μM, 0.1 μM, and 1 μM exerted no significant effect on cell viability, which were suitable for subsequent functional experiments. |
|
Animal experiment: |
|
|
Animal models |
SD rats |
|
Dosage form |
Rat Aortic Ring Assay: No in vivo administration was performed. Rat aortic rings were cultured in vitro, and anlotinib at concentrations of 0.01 μM, 0.1 μM, and 1 μM was added, together with 50 ng/mL of VEGF/PDGF-BB/FGF-2 to induce microvessel sprouting. |
|
Results |
Aortic Ring Angiogenesis: Anlotinib inhibited microvessel sprouting in a concentration-dependent manner. At a concentration of 1 μM, its inhibition rate was 15%–30% higher than that of control drugs such as sunitinib, and it exerted significant inhibitory effects on VEGF/PDGF-BB/FGF-2-induced sprouting. |
|
References: |
[1] Lin B, Song X, Yang D, Bai D, Yao Y, Lu N. Anlotinib inhibits angiogenesis via suppressing the activation of VEGFR2, PDGFRβ and FGFR1. Gene. 2018 May 15;654:77-86. doi: 10.1016/j.gene.2018.02.026. Epub 2018 Feb 14. Erratum in: Gene. 2020 Jan 10;723:144119. doi: 10.1016/j.gene.2019.144119. PMID: 29454091. |
|
The IC50 of anlotinib on kinase activity |
|||
|
Targets |
VEGFR2 |
PDGFRβ |
FGFR1 |
|
IC₅₀ |
5.6±1.2 nM |
8.7±3.4 nM |
11.7±4.1 nM |
Quality Control & MSDS
- View current batch:
-
Purity = 98.00%
- COA (Certificate Of Analysis)
- MSDS (Material Safety Data Sheet)
Chemical structure